

Improving a Fundamental Method for Comparative Genomics: Introducing OrthoHMM
Enhanced Ortholog Inference Using Hidden Markov Models for Improved Genomic Comparisons

Subscribe to Genomely for the latest discoveries and in-depth analyses in your inbox
Thank you for subscribing and for your continued support and passion for science!
A quick note
This blog post describes a new preprint myself and a team of scientists more talented than I got to publish. We are excited to share this article and the software described therein with you.
If this article piques your interest, consider reading the full technical report OrthoHMM: Improved Inference of Ortholog Groups using Hidden Markov Models, Steenwyk et al. (2024), bioRxiv.
Lastly, thank you so much for being here. I’m glad you decided to visit :)
Accurately identifying orthologous groups of genes — which serve as proxies for gene families — is fundamental for understanding functional and evolutionary relationships in comparative and evolutionary genomics. However, the task is notoriously challenging, particularly as sequence divergence increases with evolutionary time. Our recent manuscript introduces OrthoHMM, a novel algorithm that leverages Hidden Markov Models (HMMs) to improve the inference of ortholog groups. This work represents an advancement in the toolkit available to comparative genomics researchers and paves way for incorporating HMMs into gene family inference.
The Challenges of Ortholog Inference
Orthologous genes, or genes derived from a common ancestor, are foundational to genomic studies across taxa. However, traditional methods often falter in cases of extreme sequence divergence or complex evolutionary events like gene duplication and loss. Current tools often rely on network-based clustering algorithms informed by sequence similarity but often lack the sensitivity for robust inference in challenging datasets.
OrthoHMM: A Novel Approach

OrthoHMM introduces a unique method that parameterizes HMMs using amino acid substitution matrices, bypassing the limitations of traditional approaches to build HMMs, but leverages the advances offered by HMMs. By employing probabilistic modeling, OrthoHMM excels in detecting remote homologs, making it particularly adept at handling anciently diverged organisms or rapidly evolving gene families.
To clarify what the benefits of HMMs during sequence similarity search, here is a side-by-side comparison of HMM-based search, compared to a more traditional method like BLAST.
1. Sensitivity to Sequence Divergence
HMMs: Use probabilistic models, which help capture subtle patterns of conservation and variation in a single sequence, offering a richer representation of sequence variation. This allows HMMs to identify distant homologs with higher accuracy.
BLAST: Relies on pairwise alignment and sequence similarity scoring, which can miss relationships when sequences are too divergent. This is partly driver by comparing individual sequences without leveraging the evolutionary context provided by profiles.
2. Incorporation of Substitution Patterns
HMMs: Allow customization with substitution matrices like BLOSUM or PAM, which can be optimized for different evolutionary distances, improving sensitivity.
BLAST: It uses substitution matrices, but its pairwise alignment lacks the probabilistic depth of HMMs.
3. Context Awareness
HMMs: Assess the likelihood of a sequence alignment within the context of the entire model, providing a global assessment of similarity.
BLAST: Focuses on local alignments, which can sometimes miss the bigger picture of sequence relationships.
4. Utility in Multi-Sequence Scenarios
HMMs: Shine when many sequences must be compared, such as identifying protein families or orthologous groups, as they incorporate the collective information from all sequences in a group.
BLAST: Primarily optimized for pairwise comparisons, making it less effective in multi-sequence contexts.
5. Accuracy in Remote Homology
HMMs: Excel in detecting remote homologs by using profiles that better represent sequence variation across evolutionary distances.
BLAST: Struggles with remote homology due to its reliance on direct sequence similarity metrics.
OrthoHMM leverages these and other advantages offered by HMMs, thereby introducing HMMs into ortholog group inference for the first time. Moreover, OrthoHMM is easy to use; the OrthoHMM software automates the entire workflow, from all-by-all sequence comparisons to clustering genes into orthologous groups. Users can customize parameters like e-value thresholds, substitution matrices, and Markov clustering inflation values.
Benchmarking Against the Best
To validate OrthoHMM's performance, the team benchmarked it against widely-used tools like OrthoFinder, Proteinortho, OrthoMCL, and InParanoid. Two datasets were used: the established OrthoBench dataset of bilaterian orthogroups and the newly constructed Three Kingdoms dataset spanning animals, plants, and fungi.
Key results included:
Superior Precision: OrthoHMM outperformed competitors, showing a 10.3–138.9% improvement in precision.
Consistent High Performance: It excelled across both datasets, with the BLOSUM62 and BLOSUM90 substitution matrices delivering the highest accuracy.
Adaptability: While other tools struggled with complex multi-copy orthogroups or distant phylogenetic relationships, OrthoHMM maintained robust performance across scenarios.

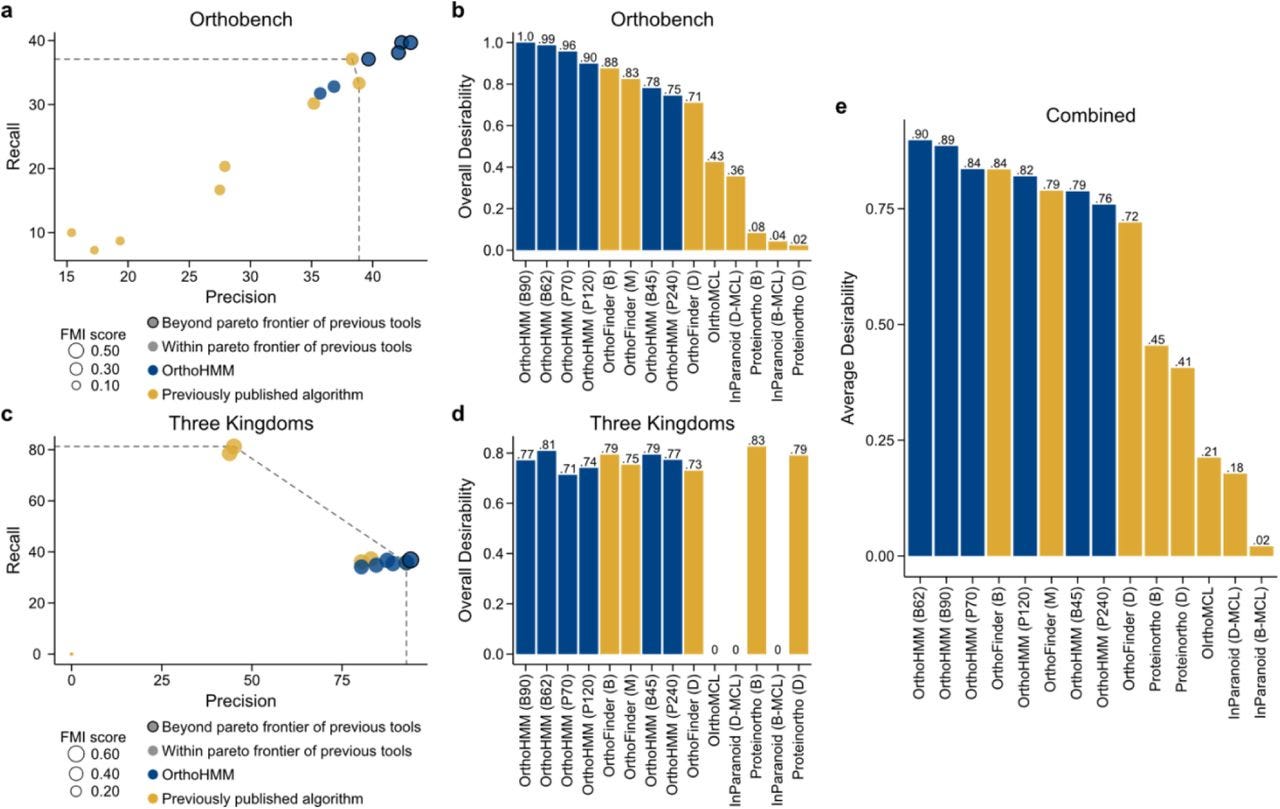
The figure above summarizes results comparing OrthoHMM to other tools. (a) Examination of various algorithms across the three-performance metrics—precision (x-axis), recall (y-axis), and Fowlkes-Mallows Index (FMI) score (data point size)—using the OrthoBench dataset revealed four variants of OrthoHMM outperformed other algorithms. (b) Desirability-based ranking, which aggregates precision, recall, and FMI score, revealed OrthoHMM using the BLOSUM90, BLOSUM62, PAM70, and PAM120 substitution matrices outperformed all other approaches. (c) Examination of the same performance metrics in the Three Kingdoms dataset revealed that one variant of OrthoHMM surpassed the pareto frontier of previous tools. (d) Desirability-based ranking corroborated OrthoHMM is a top-performing algorithm. (e) The average desirability scores in the OrthoBench and Three Kingdoms datasets revealed OrthoHMM with the BLOSUM62 and BLOSUM90 substitution matrices outperformed all other approaches. In panels a and c, the pareto frontier of previous tools is represented as a dashed line. In panels b, d, and e, the abbreviations in the paratheses provide additional information about orthology inference parameters and are as follows: B45, BLOSUM45; B62, BLOSUM62; B90, BLOSUM90; P70, PAM70; P120, PAM120; P240, PAM240; B, BLAST; D, DIAMOND; D-MCL, DIAMOND and Markov clustering; and B-MCL, BLAST and Markov clustering. OrthoHMM is depicted in blue; other algorithms are depicted in gold.
Implications for Genomic Studies
The implications of OrthoHMM extend beyond ortholog inference. Downstream analyses benefit from its accuracy, such as reconstructing gene family evolution or defining pangenomes. For instance, in analyses of Saccharomyces species and Candida auris, OrthoHMM provided refined insights into gene family gains and losses and the structure of core and accessory genomes. These examples provide small vignettes about how OrthoHMM can be used; however, scientists are a creative bunch and I anticipate others will come up with exciting applications and shed light on the fundamental principles of genome evolution.
Usability and Accessibility
OrthoHMM is not just powerful but also user-friendly. It integrates seamlessly into bioinformatics pipelines and is available as open-source software on PyPi and GitHub, complete with comprehensive documentation. The algorithm's modular design ensures long-term stability, supported by rigorous testing and a continuous integration pipeline. These industry-level standards for software development help combat an issue that plagues bioinformatics; that is, nearly 30% of all bioinformatic software fail to install, threatening the progress and reproducibility of analyses.
Looking Ahead
OrthoHMM helps pave the way for deeper insights into genome evolution. Its ability to integrate high sensitivity with flexibility ensures broad applicability, from studying deep evolutionary relationships to characterizing newly sequenced genomes.
As the research community increasingly faces the challenges of large-scale genomic datasets, tools like OrthoHMM will be instrumental to understanding life and the evolutionary processes governing it.
Congrats to you and your co-authors on developing this new tool!
Looking forward to diving into the source to learn more about the phylogenetic correction from step two in OrthoHMM workflow (i.e. "using the average reciprocal best hit score between the two taxa"). Seems like a clever way to circumvent the more computationally expensive "phylogeny-aware methods."