



A cryptic fungal pathogen hiding in plain sight
The evolutionary origin of Aspergillus latus

Subscribe to Genomely for the latest discoveries and in-depth analyses in your inbox
Thank you for subscribing and for your continued support and passion for science!
Fungal infections are a significant global health concern, affecting over 150 million people each year and resulting in an estimated 1.7 million deaths annually. The economic impact of these infections is equally devastating, with costs ranging from 11.5 billion to as high as 48 billion US dollars. The increasing incidence of fungal infections, exacerbated by global climate change and their role as co-infections with diseases like COVID-19, prompted the World Health Organization (WHO) to establish the first-ever fungal pathogen priority list. Despite recent attention, our understanding of fungal pathogenicity—especially the repeated evolution of pathogenic traits—remains limited. This is particularly true for cryptic pathogens, which are genetically distinct but morphologically indistinguishable from known species.
The Hidden Pathogen: Aspergillus latus
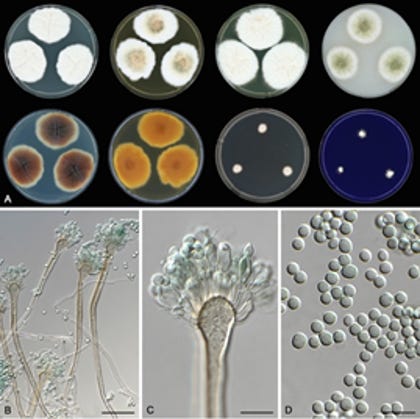
Aspergillus latus is a novel cryptic pathogen that emerged from a hybridization event between two distinct species—A. spinulosporus and an unknown close relative of A. quadrilineatus. The hybrid nature of A. latus means that it contains nearly the entire genomes of both parental species, making it an allodiploid hybrid. However, the limited number of clinical isolates studied and the use of fragmented short-read genome assemblies left many questions unanswered. To address these gaps, we performed a comprehensive analysis using high-quality long- and short-read sequencing on 53 globally distributed isolates. This deeper investigation aimed to uncover the genetic and phenotypic diversity of A. latus and how it is different from closely related species.
Expanding the Aspergillus latus Dataset
To further understand the biology and evolutionary history of A. latus, the genomes of 50 clinical isolates and three type strains were sequenced and assembled. The dataset includes isolates from patients with diverse pathologies, particularly those with lung-related diseases such as cystic fibrosis and chronic granulomatous disease. Notably, six misidentified A. latus isolates were recovered from patients co-infected with COVID-19, adding to the evidence that Aspergillus species can co-infect alongside SARS-CoV-2.
Phylogenetic analysis of taxonomically informative loci (β-tubulin and calmodulin) confirmed that the 50 clinical isolates belong to four species: A. latus, A. spinulosporus, A. quadrilineatus, and A. nidulans. Notably, A. latus was frequently misidentified using conventional methods such as matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. This misidentification likely results from the absence of A. latus in MALDI-TOF databases, suggesting that infections by this cryptic pathogen may be underreported.
The Hybrid Nature of A. latus
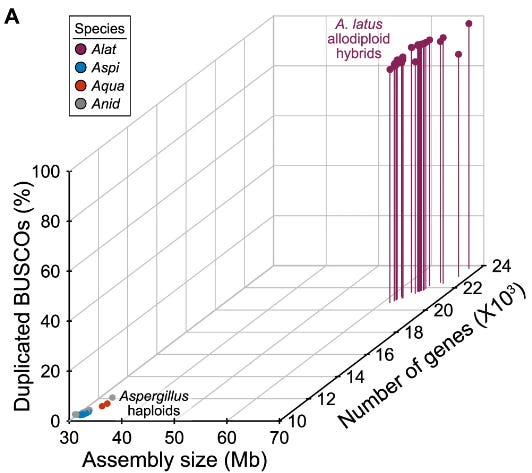
Genomic, phylogenomic, and macrosynteny analyses provided unequivocal support for the hybrid origin of A. latus. The A. latus genome is approximately twice the size and encodes nearly twice the number of genes compared to its closest relatives. Most importantly, A. latus retains duplicated copies of 96.68% of single-copy orthologs, known as ohnologs, which indicate its allopolyploid nature. Comparative analysis of these ohnologs revealed that they are evolving at indistinguishable rates, suggesting that both parental genomes are subject to similar evolutionary pressures. Further analysis showed little evidence of recombination between the parental genomes, indicating that the hybrid genome is stable and maintains the distinctiveness of both parental lineages.
Timing and Origin of A. latus

Using relaxed molecular clock analyses, we estimate that A. latus originated approximately 13.9–13.1 million years ago during the Miocene epoch. While phylogenetic topology tests initially suggested two hybridization events, further testing supported a single hybridization event as the most parsimonious explanation.
Exploring the Genetic Diversity of A. latus
Our genomic analysis uncovered substantial genetic diversity in A. latus, particularly in gene families and biosynthetic gene clusters. A total of 10,078 gene families were identified, of which 74.27% are core (present in all isolates). Among BGCs, accessory clusters (not present in all isolates) outnumber core clusters, highlighting the rapid evolution of secondary metabolic pathways in fungal hybrids. The products of these BGCs remain largely unknown, indicating that A. latus could produce novel secondary metabolites that are yet to be explored.
Phenotypic Profiles: Distinctive Traits of a Cryptic Pathogen
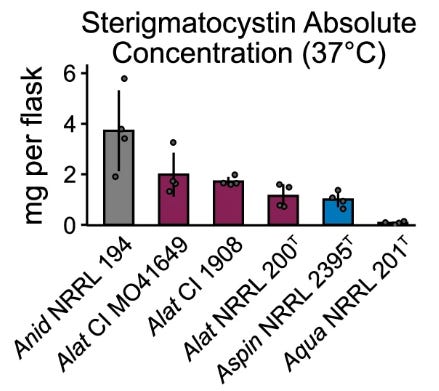
Hybridization can have profound effects on the phenotypic profiles of organisms. Extensive phenotyping of A. latus and its parental species across infection-relevant traits revealed that A. latus has a unique phenotypic profile. It differs in growth under various environmental stresses, drug resistance, spore size, and secondary metabolite production. Interestingly, A. latus hybrids produced higher levels of mycotoxin sterigmatocystin than either parental species, suggesting that hybridization may enhance certain phenotypic traits.
Additionally, RNA-sequencing analysis showed that both parental subgenomes are actively expressed and respond to environmental perturbations in potentially a coordinated manner. Approximately equal transcript abundances were observed in both subgenomes at different temperatures, indicating no strong transcriptional bias favoring one parent.
Taxonomic Identification: Towards Better Diagnostic Tools

The misidentification of A. latus using traditional diagnostic tools underscores the need for improved taxonomic methods. Our study identified several genomic and phenotypic traits that can reliably distinguish A. latus from its closest relatives. At the genomic level, A. latus isolates have larger genomes and gene repertoires, which can be detected using techniques like Fluorescence-Activated Cell Sorting (FACS) analysis of DNA content. Moreover, amplification and sequencing of single-locus molecular markers can reveal the presence of two distinct loci that are phylogenetically separate in a single-locus phylogeny. At the phenotypic level, the larger spore size of A. latus is a distinguishing feature.
Concluding Remarks and Future Directions
Our study sheds light on the evolutionary history, genetic diversity, and unique phenotypic profile of the cryptic fungal pathogen Aspergillus latus. As a hybrid species, A. latus maintains the distinctiveness of its parental genomes while exhibiting a complex interplay of additive and non-additive effects. The findings emphasize the importance of hybridization in the evolution of fungal pathogenicity and call for a re-evaluation of diagnostic methods to accurately identify and study cryptic pathogens. Future studies focusing on chromosome-level assemblies and discovering the unknown parental species will further elucidate the evolutionary dynamics and pathogenic potential of A. latus.
In conclusion, the comprehensive characterization of A. latus presented here sets the stage for future research into the epidemiology, pathogenicity, and management of this cryptic yet impactful fungal pathogen. Understanding the complex interactions between its hybrid genome and phenotypic traits will ultimately aid in developing better diagnostic tools and therapeutic strategies to combat fungal infections globally.
Reference: https://www.nature.com/articles/s41467-024-52639-1